")

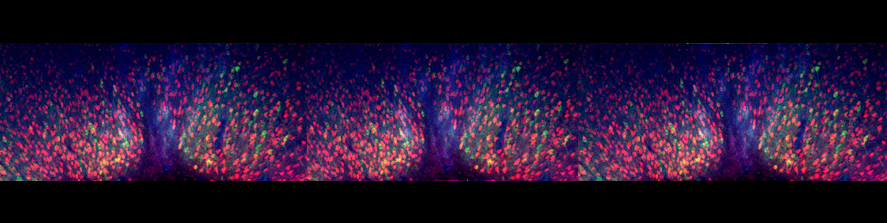
Neuroinflammation and selective vulnerability of the Entorhinal Cortex in models of neurodegeneration
The medial temporal lobe is also central to the pathophysiology of AD since histopathology suggests it is affected during the earliest phases of the disease. Specifically, abnormal protein aggregates such as amyloid plaques and neurofibrillary tangles have been observed to accumulate in this region and the density of these lesions correlates with AD severity. Within the medial temporal lobe, the distribution of the typical AD histopathological findings follows a specific progression, first beginning in layer II of the EC and then spreading to the hippocampal formation. The selective vulnerability of EC neurons might be the result of a particular susceptibility to microenviromental factors, such as increased Aβ levels, tau hyperphosphorylation vascular injury that interact with molecular peculiarities of these cells. Our studies can be summarized as follow:
- a) Role of microglia in Aβ mediated synaptic dysfunction: the role of extracellular vesicles.
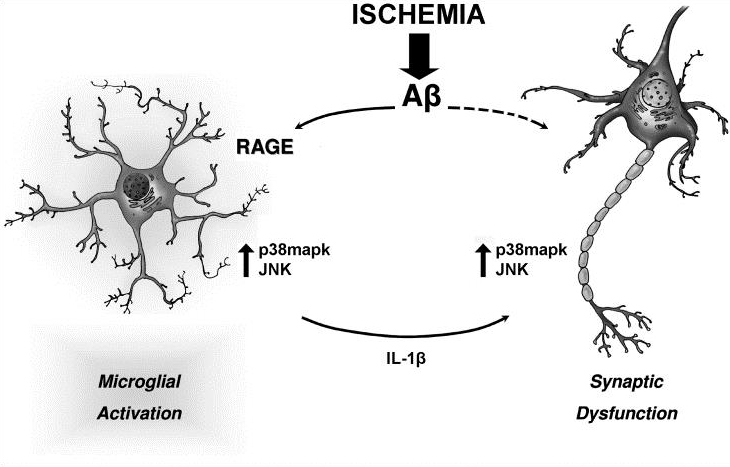
We raised the key question of whether brain neuroinflammation is involved in progressive synaptic and cognitive deficits induced by Aβ load. It is known that high Aβ concentrations induce specific cell-stress signaling and activation of proinflammatory cytokines, such as Interleukin-1β (IL-1β), leading to impaired synaptic transmission and plasticity in the EC (Origlia et al., J Neurosci 2010). The progression of synaptic dysfunction by Aβ is concentration dependent, possibly corresponding to cognitive decline induced by its accumulation during AD. We confirmed the temporal profile of Aβ-dependent synaptic and behavioral dysfunctions in mutant human APP transgenic mice (J20 line), characterized by progressive accumulation of Aβ (Criscuolo et al., 2017). However, how synaptic dysfunction originates and propagates in the affected brain is still largely obscure, and it is now one of the most compelling questions in Alzheimer’sdisease research

We have demonstrated that the motion of microglia-derived large extracellular vesicles carrying β-amyloid, at the neuronal surface, is crucial for the initiation and propagation of synaptic dysfunction along the entorhinal–hippocampal circuit. Using chronic EEG recordings, we show that a single injection of extracellular vesicles carrying β-amyloid into the mouse entorhinal cortex could trigger alterations in the cortical and hippocampal activity that are reminiscent of those found in Alzheimer’s disease mouse models and human patients. The development of EEG abnormalities was associated with progressive memory impairment as assessed by an associative (object-place context recognition) and non-associative (object recognition) task. Importantly, when the motility of extracellular vesicles, carrying β-amyloid, was inhibited, the effect on network stability and memory function was significantly reduced. Our model proposes a new biological mechanism based on the extracellular vesicles–mediated progression of β- amyloid pathology . Gabrielli et al., 2022; Falcicchia et al., 2023).
b). Vascular and metabolic factors implicated in Aβ-dependent EC dysfunction
Hypoxia may be a cause of the progressive neuronal alterations in AD. However, a causal relationship between oxygen deficiency and AD at the cellular and molecular level has not been established We addressed the hypothesis that transient ischemia facilitates EC synaptic impairment induced by Aβ signaling. We demonstrated that transient brain hypoxia/ischemia functions as a trigger for neuronal perturbation induced by progressive accumulation of Aβ and that microglia-mediated inflammation important factor in accelerating synaptic dysfunction (Origlia et al; 2014). Recently we focused on alternative strategies to interfere with Aβ signaling in the attempt to reduce synaptic dysfunction. In particular, we investigated the role of thyroid hormones in the brain, as their availability and/or metabolism have been hypothesized to contribute to Alzheimer’s disease (AD) and to be a risk factor for stroke. The 3-iodothyronamine (T1AM) metabolite, an endogenous amine putatively derived from TH metabolism, gained interest for its ability to promote learning and memory in the mouse. Moreover, we demonstrated that T1AM and its putative receptor TAAR1 are part of an endogenous system that can be modulated to rescue the β-Amyloid dependent EC dysfunction (Accorrroni et al., 2020). Then, we confirm that T1AM can rescue synaptic function after transient ischemia in the EC, an effect that was also observed in a Aβ-enriched environment (Tozzi et al., 2021). Since T1AM and synthetic analogues were shown to be effective in ameliorating neurodegeneration , these compounds and other TAAR1 agonists may represent a novel strategy for neuroprotection.